Reakcja SN1

SN1 – uniwersalnie stosowany w chemii organicznej symbol dla reakcji chemicznej substytucji nukleofilowej zachodzącej poprzez mechanizm jednocząsteczkowy.
Reakcje te polegają na wymianie atomu lub grupy atomów na inne pod wpływem działania nukleofila, przy czym etapem kluczowym (decydującym o szybkości i kierunku procesu) jest oderwanie od centralnego atomu grupy opuszczającej z utworzeniem mniej lub bardziej trwałego kationu.
Mechanizm ten został pierwotnie zaproponowany przez Christophera Ingolda w roku 1940[1].
Alternatywnym mechanizmem przebiegu organicznych reakcji substytucji jest SN2. To czy reakcja zachodzi według mechanizmu SN1 lub SN2 zależy od warunków reakcji oraz od substratów. Czasami zdarza się też, że ta sama reakcja zachodzi jednocześnie poprzez oba mechanizmy. W wyniku reakcji substytucji zachodzącej zgodnie z mechanizm SN1 przy chiralnym atomie węgla powstaje mieszanina racemiczna. Zazwyczaj jednak racemizacja nie jest całkowita, prawdopodobnie w wyniku tworzenia pary jonowej pomiędzy grupą opuszczającą a powstałym karbokationem, w efekcie czego jedna jego strona jest lepiej dostępna dla atakującego nukleofila[2].
Mechanizm[edytuj | edytuj kod]
Przykładem reakcji SN1 jest hydroliza bromku tert-butylowego w wyniku której powstaje alkohol tert-butylowy, anion bromkowy i jon hydroniowy[3]:

Reakcja SN1 zachodzi w trzech etapach, które są zilustrowane na przykładzie reakcji bromku tert-butylu z wodą:
- 1. Tworzenie karbokationu: W wyniku oderwania jonu bromkowego powstaje płaski karbokation tert-butylowy. Etap ten jest najwolniejszy. To on decyduje o szybkości całej reakcji:

- 2. Atak nukleofila: Karbokation reaguje z nukleofilem, w tym przypadku z cząsteczką wody. Nukleofil może zaatakować karbokation z obu stron z równym prawdopodobieństwem, co jest przyczyną racemizacji w przypadku reakcji SN1 związków chiralnych. Jeżeli nukleofil jest anionem, na tym kończy się reakcja, jeśli nie jest, powstaje kolejny produkt przejściowy, w tym przypadku kation t-butylohydroksoniowy:

- 3. Deprotonacja: Reakcję w takim przypadku kończy trzeci etap zwany deprotonacją, polegający na oderwaniu się protonu i powstaniu alkoholu oraz jonu hydroniowego.
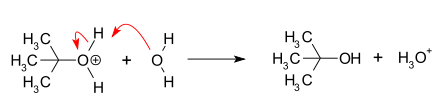
- Deprotonacja również zachodzi z względnie dużą szybkością.


Wszystkie etapy reakcji SN1 są odwracalne. W obecności wody równowaga przesunięta jest w kierunku alkoholu, natomiast w środowisku bezwodnym alkohol tert-butylowy reaguje z bromowodorem z wytworzeniem bromku tert-butylowego[4].
Kinetyka[edytuj | edytuj kod]
W przeciwieństwie do reakcji typu SN2, reakcja SN1 przebiega w dwóch etapach (nie licząc protonacji czy deprotonacji). O szybkości całej reakcji decyduje etap pierwszy, w którym tworzy się karbokation w wyniku dysocjacji wiązania C–X (gdzie X = halogen, OH, SH lub NH). Etap ten jest bowiem najwolniejszy. Reakcja SN1 jest reakcją pierwszego rzędu (jednocząsteczkową), gdyż produkt pośredni tworzy się wyłącznie z cząsteczki substratu. Oznacza to, że jej szybkość zależy jedynie od stężenia substratu, a nie stężenia nukleofila:
- r=kCs
gdzie:
- r – szybkość reakcji
- k – stała szybkości reakcji
- Cs – stężenie substratu organicznego RX
Przypisy[edytuj | edytuj kod]
- ↑ Leslie C. Bateman i inni, Mechanism of Substitution at a Saturated Carbon Atom. Part XXIII. A Kinetic Demonstration of the Unimolecular Solvolysis of Alkyl Halides. (Section E) A General Discussion, „Journal of the Chemical Society”, 1940, s. 979–1011, DOI: 10.1039/JR9400000979.
- ↑ John McMurry: Chemia organiczna. Wyd. 3. T. 2. Warszawa: PWN, 2005, s. 388. ISBN 83-01-14406-8.
- ↑ Robert T. Morrison, Robert N. Boyd: Chemia organiczna. T. 1. Warszawa: PWN, 1985, s. 544–550. ISBN 83-01-04166-8.
- ↑ Hiromitsu Masada, Yoshiharu Murotani, A Convenient Method for the Preparation of Highly Pure t-Alkyl Bromides and Iodides, „Bulletin of the Chemical Society of Japan”, 4, 53, 1980, s. 1181–1182, DOI: 10.1246/bcsj.53.1181.
Bibliografia[edytuj | edytuj kod]
- Przemysław Mastalerz: Chemia Organiczna. Wrocław: Wydawnictwo Chemiczne, 2000. ISBN 83-905776-4-X.