Chromatographie en phase gazeuse

Pour les articles homonymes, voir CPG.
La chromatographie en phase gazeuse (CPG) est une technique de chromatographie qui permet de séparer des molécules d'un mélange gazeux, éventuellement très complexe, de natures très diverses. Elle s'applique principalement aux composés gazeux ou susceptibles d'être vaporisés par chauffage sans décomposition. Elle est de plus en plus utilisée dans les principaux domaines de la chimie, ainsi qu'en parfumerie et en œnologie.
Le mélange à analyser est vaporisé à l'entrée d'une « colonne », qui renferme une substance active solide ou liquide appelée « phase stationnaire », puis il est transporté à travers celle-ci à l'aide d'un gaz porteur (ou gaz vecteur). Les différentes molécules du mélange se séparent et sortent de la colonne les unes après les autres après un certain laps de temps qui est fonction de l'affinité de la phase stationnaire avec ces molécules.
Historique[modifier | modifier le code]
En 1944, Erika Cremer met au point avec l'un de ses étudiants le premier chromatographe avec phase mobile gazeuse, afin de pouvoir séparer des gaz. Ses travaux qui établissent les bases théoriques de la technique sont publiés en 1951 dans une petite revue de langue allemande, mais le sujet est considéré comme sans intérêt par la communauté scientifique autrichienne[1]. En 1952, A.J.P Martin et R.L.M. Synge en Angleterre, et Alain Berton en France annoncèrent la naissance de la chromatographie en phase gazeuse. Cette technique a vécu son âge d'or entre 1955 et 1960, avec l'invention des colonnes capillaires par Marcel Golay (1957), du détecteur à ionisation à argon (1958), suivi du détecteur à ionisation de flamme (1958) et du détecteur à capture d'électrons (1960). Dès les années 1960, les progrès se sont orientés sur l'instrumentation et ont permis de rendre viables toutes ces inventions. De la fin des années 1970 à la fin des années 1980, d'énormes recherches ont été entreprises pour permettre l'analyse de toutes les familles de composés chimiques, grâce notamment au développement de nouveaux injecteurs et des colonnes capillaires.
Dès 1962, cette technique d'analyse est utilisée couramment dans l'industrie pétrolière. En effet, pour avoir des résultats rapides lors des forages, il est indispensable d'avoir une méthode d'analyse qui donne presque instantanément et automatiquement des résultats fiables. Depuis, cette technique s'est développée et s'étend aujourd'hui à tous les domaines : chimie, biologie, astronomie, pharmacie, industrie des matières plastiques, etc.
Compte tenu de ses nombreuses applications dans tous les domaines des sciences, la chromatographie de grande efficacité est considérée comme une évolution majeure du XXe siècle dans le domaine de la chimie analytique.
Appareils[modifier | modifier le code]

Les appareils de chromatographie gazeuse sont appelés chromatographes. Ils sont principalement composés:
- d'un four (type chaleur tournante) qui permet une programmation de température ajustable de 20 °C (−100 °C pour certains systèmes) à 450 °C et qui est également équipé d'un système de refroidissement rapide;
- d'un système d'injection, qui va permettre d'introduire et de rendre volatil l'échantillon à analyser. L'injection peut se faire d'une manière manuelle ou automatique à l'aide d'un échantillonneur;
- d'une colonne (capillaire ou remplie) qui peut faire plus de 50 mètres, sur laquelle les différentes molécules de l'échantillon injecté vont se séparer suivant leurs affinités avec la phase stationnaire;
- d'un système de détection, qui va permettre de mesurer le signal émis par les différentes molécules et de pouvoir les identifier. Pour l'enregistrement du signal émis par le détecteur, des logiciels sur PC remplacent avantageusement les enregistreurs analogiques sur papier;
- d'un système de détendeur-régulateur pour les gaz utilisés (hélium, dihydrogène, diazote et air comprimé). Sur les chromatographes modernes, on trouve des systèmes électroniques pour la régulation des gaz qui sont également purifiés par des cartouches filtrantes.
Il existe des chromatographes de différentes tailles. Cela va du portable (env. 10 kg) conçu pour les analyses sur le terrain, à ceux utilisés dans la purification des gaz rares.
Il existe trois sortes de chromatographes :
- Chromatographe industriel, utilisé pour la purification des produits, c'est ainsi qu'Air Liquide a conçu et fabriqué un chromatographe de grande taille pour la purification du krypton et du xénon.
- Chromatographe à colonne, utilise un produit (phase stationnaire) imprégné ou greffé sur un support solide inerte à forte capillarité
- Chromatographe capillaire, ou la phase stationnaire est fixée directement sur la surface interne du tube capillaire creux, dont le diamètre interne est de l'ordre du demi ou quart de millimètre.
La chromatographie de type gaz-liquide, largement utilisée de nos jours par rapport à celle de type gaz-solide, se fonde sur le partage du soluté entre une phase mobile gazeuse et une phase stationnaire liquide immobilisée sur un support inerte.
Principe de fonctionnement[modifier | modifier le code]
L'échantillon (un liquide volatil) est d'abord introduit en tête de colonne par l'intermédiaire d'une micro seringue qui va traverser une pastille souple, appelée septum, pour se retrouver dans une petite chambre en amont de la colonne appelée injecteur. L'injecteur est traversé par le gaz porteur et porté à une température appropriée à la volatilité de l'échantillon. Les quantités injectées peuvent varier de 0,2 à 5,0 μl.
Ensuite, une fois rendus volatils, les différents composés de l'échantillon vont être emportés par le gaz porteur (ou gaz vecteur) à travers la colonne et se séparer les uns des autres en fonction de leur affinité avec la phase stationnaire. La phase stationnaire peut être un liquide non (ou peu) volatil (chromatographie gaz-liquide) ou un solide adsorbant (chromatographie gaz-solide). Dans les deux cas, la phase stationnaire va provoquer un phénomène de rétention chromatographique avec les différents composés (appelés solutés). Plus le composé a d'affinité avec la phase stationnaire, plus il mettra de temps à sortir de la colonne. La grandeur expérimentale brute est appelée temps de rétention. C'est le temps qui s'écoule entre l'injection de l'échantillon et l'apparition du signal maximum du soluté au détecteur. Pour favoriser le transport de tous les composés à travers la colonne (élution), il faut déterminer la bonne température du four. En général, la température doit être légèrement supérieure à la température d'ébullition des composés (de manière que les composés ne sortent pas trop tôt, ce qui aurait pour conséquence d'avoir leurs pics confondus avec celui du temps mort). On peut travailler en isotherme, c’est-à-dire avec une température fixe durant toute l'analyse ou avec un programme de température qui varie.
À la sortie de la colonne, les composés rencontrent un élément essentiel qui est appelé détecteur. Cet élément évalue en continu la quantité de chacun des constituants séparés au sein du gaz porteur grâce à la mesure de différentes propriétés physiques du mélange gazeux. Le détecteur envoie un signal électronique vers un enregistreur de données qui dessinera les courbes de chaque pic en fonction de leur intensité (courbe de type Gaussienne). L'ensemble des pics est appelé chromatogramme. Actuellement et de plus en plus, les logiciels remplacent avantageusement les enregistreurs papiers pour l'interprétation des signaux envoyés par les détecteurs.
Notions théoriques[modifier | modifier le code]
Théorie des plateaux[modifier | modifier le code]
Afin de simplifier la représentation théorique des équilibres entre phase, les chercheurs Martin et Synge publient en 1950 leur modèle des plateaux[2]. Dans ce dernier, on considère la colonne composée de petits cylindres juxtaposés et la phase mobile se déplaçant par à-coups de l'un à l'autre. Dès lors, il devient plus aisé de calculer les équilibres entre phase stationnaire et mobile pour chacun de ces plateaux. Le nombre de plateaux est un indicateur de performance d'une colonne. Plus il est grand et plus la rétention sera importante, permettant de séparer des composés très proches. Expérimentalement, le calcul de N se fait à partir des caractéristiques , et de pic (cf. Paragraphe ci-dessous) :





On rencontre parfois aussi la Hauteur Équivalente à un Plateau Théorique (HEPT) définie comme et la plupart du temps comprise entre 0,1 et 10 mm[3].

Caractéristiques et résolutions des pics[modifier | modifier le code]

Outre le temps de rétention (lu en abscisse), plusieurs grandeurs peuvent être mesurée sur un pic. La largeur à la base , la largeur à mi-hauteur et l'écart-type . Les relations entre ces différents paramètres sont[4] :

Lorsque les composés sortent de la colonne, chacun produit un pic sur le chromatogramme. Cependant, en fonction du débit de gaz (vitesse d'élution), la séparation entre deux pics peut ne pas être optimale. Cette grandeur se nomme résolution et est considérée bonne lorsque . Elle se calcule pour deux pics et de la façon suivante :





- Exemples de résolution
-
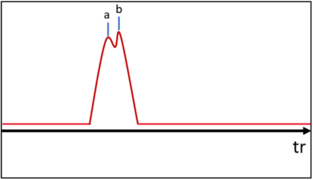 Mauvaise résolution
Mauvaise résolution -
 Moyenne résolution
Moyenne résolution -
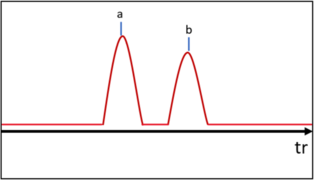 Bonne résolution
Bonne résolution



Équilibre et Rétention[modifier | modifier le code]
Le temps de rétention, notion majeure de toute technique chromatographique, est défini pour chaque composé comme la durée passée dans la colonne. Il s'agit de la grandeur permettant la séparation des différentes molécules d'un échantillon[5].
- Le temps de rétention () est la durée s'écoulant entre l'injection de l'échantillon dans la colonne et la détection du composé par le détecteur.
- Le temps mort () correspond au temps de rétention du gaz vecteur. Il s'exprime en fonction de la longueur de la colonne et de la vitesse linéaire du gaz. Ou encore en fonction du volume de la colonne et du débit de la phase mobile.
- Le temps de rétention réduit () correspond à la différence entre le temps de rétention du composé et le temps mort.









Il est possible, en suivant le même raisonnement de définir le volume de rétention (), le volume mort () ainsi que le volume de rétention réduit ().



Un équilibre existe entre le composé adsorbé sur la phase solide et dans la phase mobile : . Le rapport de phase est une grandeur caractéristique de chaque colonne, dépendant du volume de chacune des phases. La constante d'équilibre peut s'exprimer en fonction des paramètres de rétention déterminés précédemment.



![{\displaystyle {\ce {A_{(s)}<=>[K] A_{(g)}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/843e230794c6a17b45d6d1a1e3c63a205e9a5232)


![{\displaystyle K={\frac {[A_{(s)}]}{[A_{(g)}]}}={\frac {m_{s}}{m_{m}}}\cdot {\frac {V_{m}}{V_{s}}}={\frac {m_{s}}{m_{m}}}\beta }](https://wikimedia.org/api/rest_v1/media/math/render/svg/9928001e1d5bcda2cf81e02ffa70e29fdb1ea236)


Gaz[modifier | modifier le code]
Le gaz porteur (ou gaz vecteur), est la phase mobile, dynamique de la chromatographie en phase gazeuse. C'est dans son flux que l'on injecte le mélange à analyser, et c'est lui qui le véhicule jusqu'au détecteur à travers toute la colonne.
Dans la plupart des cas, il doit être inerte vis-à-vis des solutés et de la phase stationnaire. Quatre types de gaz sont surtout utilisés : hélium, dihydrogène, diazote et argon[7]. Ils peuvent être fournis soit par des cylindres de gaz ou produits par des générateurs (cas du dihydrogène et du diazote). Ces gaz vecteurs se doivent d'être purs, exempts d'eau, de dioxygène et d'hydrocarbures légers pour éviter toutes réactions avec les solutés et la phase stationnaire. C'est pourquoi des filtres spécifiques sont apposés à l'entrée du chromatographe.
La principale propriété des gaz vecteurs est leur insolubilité dans les liquides. Leur signal électrique n'apparaîtra pas sur le chromatogramme.
Injecteurs[modifier | modifier le code]
L'injecteur est logé dans un bloc métallique dont la température est régulée afin d'assurer une bonne homogénéité thermique du système. L'échantillon va être introduit, à travers une pastille auto-obturante appelée septum, par l'intermédiaire d'une micro seringue. L'échantillon sera vaporisé et les solutés traverseront l'injecteur à travers un tube en verre (parfois métallique) appelé liner (ou insert), grâce au gaz porteur, jusqu'à la tête de la colonne. L'intérêt du liner est de retenir les constituants non volatils de l'échantillon, impropres par nature à la chromatographie.
Il existe deux types d'injecteurs. Ceux pour les colonnes remplies (peu nombreux actuellement) et ceux pour les colonnes capillaires (le plus fréquent). Le principe reste le même, c'est juste une question de conception de la chambre de vaporisation ainsi que le raccordement à la colonne qui change.
Dans le cas des colonnes capillaires, trois modes d'injections peuvent se présenter:
- injection avec division (split): L'injection en split est utilisé pour les échantillons concentrés. l'échantillon est vaporisé et mélangé dans le gaz porteur, puis le mélange est divisé en deux parties. La plus petite partie arrive sur la colonne alors que la plus importante est évacuée. On l'appelle la fuite. Le ratio de la division se règle sur la machine. Ce mode d'injection permet d'injecter de petites quantités d'échantillons concentrés sans devoir les diluer au préalable. Dilution qui est parfois impossible pour certains produits (huiles essentielles, produits pétroliers...) car le solvant masquerait la détection des composés les plus volatils. Par contre, il est souvent nécessaire d'utiliser des températures d'injections élevées qui pourraient conduire à la dégradation de certains solutés.
- injection sans division (splitless) : l'échantillon est vaporisé et mélangé dans le gaz porteur, mais le mélange n'est pas divisé en deux parties. Il reste quelques secondes dans le liner avant d'être transféré sur la colonne (environ 95 % du produit). Le 5 % restant est évacué par l'ouverture de la vanne de fuite. Cette méthode est utilisée quand l'échantillon à analyser est très dilué et éventuellement très sale (contenant des résidus non-volatils). Elle permet également d'analyser les composés très volatils (plus volatils que le solvant de dilution) en les concentrant sur la tête de la colonne qui sera plus froide que l'injecteur.
- injection dans la colonne (on-column) : il n'y a pas d'étape de vaporisation. L'échantillon est directement mélangé au gaz vecteur et injecté à froid sur la colonne. Cette méthode nécessite une seringue et un injecteur spécifique (sans septum et sans chauffage). Les avantages sont de pouvoir injecter l'échantillon sous forme liquide sans provoquer de vaporisation sélective dans l'aiguille (plus de précision sur le volume d'injection) et à une température la plus basse possible (celle de la colonne) pour éviter la dégradation des composés thermolabiles. Les effets indésirables du septum (traces de polymères emportés par le gaz porteur) sont également éliminés. Par contre, des inconvénients comme l'accumulation de composés non volatils dans la colonne peuvent se présenter et la fiabilité de cette technique n'est pas toujours au rendez-vous.
Colonne[modifier | modifier le code]

La colonne est placée dans un four pour maintenir une température suffisante afin de garder les solutés en phase gazeuse pendant l'analyse.
La colonne est constituée d'un tube plus ou moins long (qui peut être en silice, acier inoxydable...) garni d'un support solide inerte (comme un zéolite) et en particules assez fines. Ce support est imprégné chimiquement d'un produit appelé phase stationnaire dont l'affinité avec les composants du produit à analyser est la plus grande. En injectant un échantillon à l'entrée de la colonne, le produit est vaporisé par chauffage et la différence d'affinité des composants envers la phase stationnaire permet de retenir plus ou moins longtemps certains composants vis-à-vis des autres. Le gaz porteur va véhiculer ces composants vers la sortie.
Il existe deux types de colonne : remplie (maximum 2 mètres) et capillaire (de 15 à 100 mètres). La différence entre celles-ci est due au type de phase stationnaire qui y est contenue : pour les colonnes remplies, ce sont des grains de silice sur lesquels repose un film liquide alors que pour les colonnes capillaires le film est directement déposé sur les parois de la colonne. Le film peut être simplement déposé ou greffé. S'il est nécessaire d'atteindre des températures élevées, on optera pour la greffe, question de stabilité.
En général, la perte de charge est assez grande entre l'entrée et la sortie de la colonne, aussi une certaine pression est appliquée pour que le gaz porteur puisse acheminer les différents composants vers la sortie.
Si l'échantillon à analyser est un mélange de gaz (dioxygène, diazote, méthane, éthane…), afin de retarder la progression de ces constituants dans la colonne, celle-ci est réfrigérée à l'extrême, on la met dans de le diazote liquide qui bout à −196 °C.
Détecteur et enregistreur[modifier | modifier le code]
À la sortie de cette colonne, un détecteur très sensible est placé, par exemple :
- Un TCD : détecteur à thermo-conduction, basé sur le principe du pont de Wheatstone : le passage des composants va faire varier la tension, cette variation est due à la différence de conductibilité de chaque composant.
- Un FID : détecteur à ionisation de flamme : une tension de l'ordre de la centaine de volts est maintenue entre la buse de la flamme et une électrode entourant cette dernière. Lorsque les molécules traversent la flamme, elles sont ionisées ce qui provoque entre les électrodes un courant électrique qui est ensuite amplifié.
- Un ECD : détecteur à capture d'électrons : des électrons sont émis, en général par une source radioactive (rayonnement bêta), et traversent le gaz ; lorsqu'un électron rencontre une molécule de gaz, il peut être capturé, ce qui fait varier l'intensité du courant d'électrons, cette intensité étant mesurée en continu.
- Un NPD : détecteur azote-phosphore connu aussi sous le nom de détecteur thermoïonique spécifique ou DTS.
- Un AED : détecteur d'émission atomique.
- Un ECD : détecteur à capture d'électrons.
- Un FPD : détecteur à photométrie de flamme.
- Un PID : détecteur à photo-ionisation.
- Un MS : spectromètre de masse, utilisant principalement l'ionisation électronique (anciennement appelée impact électronique à tort car il n'y a pas d'impact mais une déformation du nuage électronique) ou l'ionisation chimique comme modes d'ionisation.

À l'heure actuelle, il existe des détecteurs ultra sensibles permettant de détecter quelques ppm (parties par million) d'un composant. On enregistre cette variation sur l'enregistreur en fonction du temps de sortie du pic, dit temps de rétention. Les appareils actuels sont couplés avec un ordinateur. La réponse du détecteur est enregistrée dans un fichier stocké sur le disque dur et affichée simultanément en temps réel sur l'écran de l'ordinateur. Cet enregistrement constitue le chromatogramme.
La nature des composants est donnée par le temps au bout duquel apparaît le pic (temps de rétention). Pour mettre en relation le temps et la nature chimique, on se sert d'un échantillon de référence. À la sortie, le chromatogramme va fournir une série de pics plus ou moins séparés, plus ou moins grands et plus ou moins larges. La surface d'un pic est, suivant la méthode de détection, proportionnelle à la quantité de produit représentée par ce pic. En mesurant la surface de chaque pic et en la rapportant à la surface totale de tous les pics, on détermine le pourcentage de chacun des composants contenus dans le mélange analysé (méthode de mesure par normalisation). Au début, en 1955, ce calcul se faisait à la main (soit à l'aide d'un planimètre, soit par découpage du pic suivi de la pesée du papier découpé. Dans les des années 1970 cette mesure se faisait de manière automatique par des intégrateurs mécaniques puis électroniques. Actuellement, l'analyse du chromatogramme (détermination des temps de rétention et de la surface des pics) se fait à partir du fichier enregistré par un programme informatique dédié.
En parfumerie et en œnologie, le nez humain est aussi utilisé, en tant que détecteur extrêmement sensible à certaines molécules odorantes. Pour cela, une partie du flux gazeux en sortie de colonne est refroidi et humidifié avant d'être dirigé vers un cône nasal qui permet à l'opérateur de percevoir l'odeur du ou des composés séparés. Cet équipement est appelé GC-olfactomètre ou GCO.
Références[modifier | modifier le code]
- Leslie S. Ettre, « The Beginnings of Gas Adsorption Chromatography 60 Years Ago », LCGC North America, vol. 26, no 1, 1e janvier 2008, p. 48–60 (lire en ligne).
- A. H. Gordon, A. J. Martin et R. L. Synge, « Partition chromatography in the study of protein constituents », The Biochemical Journal, vol. 37, no 1, , p. 79–86 (ISSN 0264-6021, PMID 16747604, PMCID 1257847, DOI 10.1042/bj0370079, lire en ligne, consulté le ).
- (en) Pauline M. Doran, « Chapter 11 - Unit Operations : Theoretical Plates in Chromatography », dans Bioprocess Engineering Principles (Second Edition), Academic Press, (ISBN 978-0-12-220851-5, DOI 10.1016/b978-0-12-220851-5.00011-3, lire en ligne), p. 445–595.
- Marie Paule Basset, « Notions fondamentales de chromatographie »
 [PDF], sur chemphys.fr (consulté le ).
[PDF], sur chemphys.fr (consulté le ). - « theorie chromato », sur www.masterchimie1.universite-paris-saclay.fr (consulté le ).
- Marie Paule Bassez, « Notions fondamentales de chromatographie » [PDF] (consulté le ).
- Gwenola Burgot, Jean-Louis Burgot, Méthodes instrumentales d'analyse chimique et applications, Lavoisier, 3e édition, 2011.